|
Flyguy
User Login
Dictators
Redbrick
Paranoid?
Harry Manback
Downloads
Robert Joyce
Bad Jokes
Bleeding Machine
fairies
Dead Kennedys
The Noble Cow
Genomic Dotplot
Cobra
Reading List
Origin
users
logged in
|
Pairwise Sequence Alignment
Firstly there is no universally precise, applicable notion of similarity, rather we choose the best
technique for each instance. An alignment is an arrangment of sequences, which highlights where the
sequences are similar and where they differ. From this it is obvious that the optimal alignment is
the arrangement which exhibits the most important similarities and least differences.
There are three extensively used methods in sequence alignment:
-
Segment Methods
- all windows are iteratively compared against the match sequence. The window is
a predetermined size, Dotplots use this approach
-
Optimal Global Alignemnt
- the best global match is found for the entire sequence, taking gaps into
consideration. This is a highly specific technique and may lead to erroneous results for large sequences
-
Optimal Local Alignment
- this algorthim searches for the best local alignment, explicitly taking gaps into
consideration.
Dotplots
Dotplots provide an intuitive representation of the comparison between two sequences, making them one of most commonly
used graphical techniues. The two sequences are represented on the X-Y axis, where significant matches are represented
on the diagonal of this matrix. Mismatches vary away from the main diagonal, to what degree depends on the exhibitted
differences.
Aside from the two sequences there are two main parameters which affect the representation of the dotplot
-
Window Size - this defines how many elements (genetic code in this case) we should try match in each comparison.
The large the window size is, the more stringent the requirement for a match are.
-
The Threshold - the threshold size dictates how many mismatches can be tolerated before we classify this comparison
as a mismatch. Thus as we increase our window size we would also expect the threshold to increase to prevent our
match requirements from becoming too stringent.
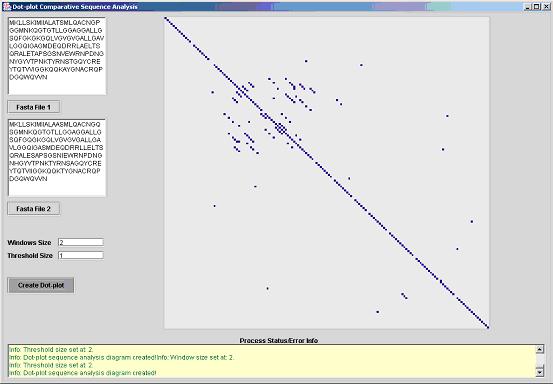
The fist image from the following two screenshots depicts how a large
threshold in relative terms of a small window can lead to the toleration of more
partial & accidental matches. Large threshold and small windows are recommended
in cases where the similarity may be weak or the genome has undergone extensive
mutations.
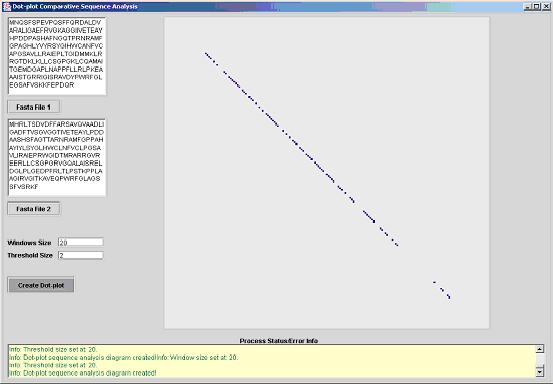
The second image is a case where there is a small threshold and a large window,
the basic laws of probability dictates that the chances of accidental matches
are very low. This "unforgiving" arrangement between the threshold and the window
size is used in cases where the hypothesis for similarity is strong, or in case
where there are similar genomes mutating slowly.
|
|
|
Fig 1. Dot-plot with a high threshold and a small window.
|
Fig 2. Dot-plot with a comparatively low threshold in relation
to a large window.
|
FASTA Formatted Sequences Files
Below are the FASTA format files that were used in this comparison. They are from the
-
Agrobacterium Tumefaciens
- a soil bacterium which infects host plants with its DNA.
Agrobacterium tumefaciens is a species of bacteria that causes tumors
(commonly known as 'galls' or 'crown galls') in dicots.
This Gram-negative bacterium causes crown gall by inserting a small segment
of DNA (known as the T-DNA, for 'transfer DNA') into the plant cell,
which is incorporated at a semi-random location into the plant genome.
These properties enable reasearchers to use this bacterium to deliver
foreign DNA into plants.
FASTA File
-
Rickettsia Prowazekii
- Epidemic Typhus is a form of typhus caused by the Bacillus Rickettsia Prowazekii, carried by
the human body louse Pediculus Humanus. Feeding on a human who carries the bacillus infects the louse. R. prowazekii
grows in the louse's gut and is excreted in the feces. The disease is transmitted to an uninfected human who scratches
the bite and rubs the feces into the wound. Incubation period is one to two weeks.
FASTA File
-
Sinorhizobium meliloti - is a nitrogen-fixing bacterium (rhizobia). It forms a symbiotic relationship with legumes from
the genera Medicago, Melilotus and Trigonella, including model legume Medicago truncatula. The S. meliloti genome
contains three replicons: a 3.65 megabase chromosome and two megaplasmids, pSyma (1.35 megabases) and
pSymb (1.68 megabases), that have all been fully sequenced.
FASTA File
What Sequences Were Compared?
For this example, I have done a quick comparison of the Rickettsia Prowazekii Genome to the Rickettsia Conorii genome
to show areas where they differ and where they correlate. Also included is how altering the window size and threshold
affects the results of the Dotplot.
Comparison of the Rickettsia Prowazekii Genome to the Rickettsia Conorii
Java Dot-Plotter
The following program was created through Java to implement pair-wise comparisons on FASTA formatted sequences. From the
UI one is able to choose the threshold and window sizes. The source file are included below in PDF format.
DotPlot.java
- Computes the co-ordinates of the dot-plot, thresolding and windowing.
DotPlotUI.java
- Creates the user interface for the dot-plot.
GraphPane.pdf
- Displays the dot-plot in the grphical pane.
|